‘Prion’ is a term first used to describe the proteinaceous infectious agent responsible for several neurodegenerative diseases found in mammals and humans. The word is derived from ‘proteinaceous infectious particle’. Prions consist only of protein, with no nucleic acid genome. Before prions were identified, diseases such as Creutzfeldt-Jakob disease and other spongiform encephalopathies were thought to be caused by viruses.
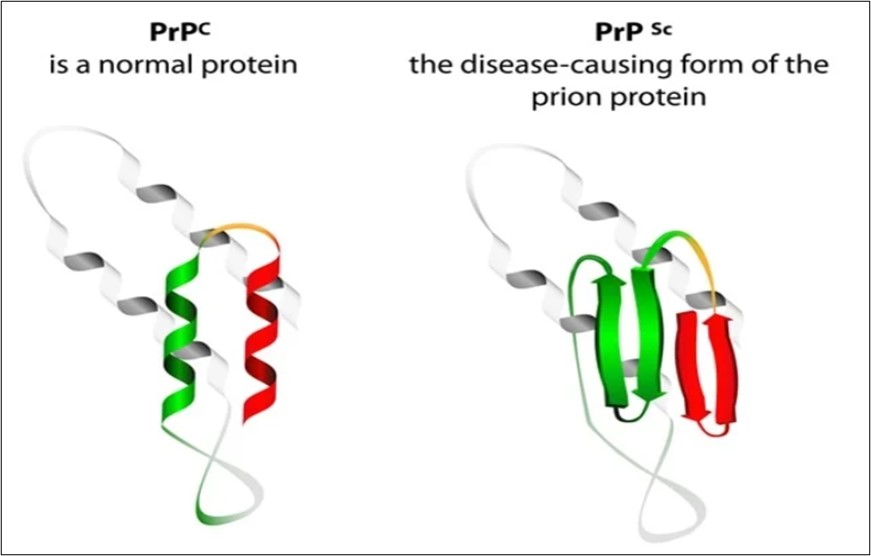
Prion diseases have attracted enormous attention for their unique biological features. Prion diseases are rare progressive, fatal neurodegenerative diseases of humans and animals as they pose threats to public health and exemplify a novel infection mechanism. In prion diseases, prions trigger normal protein to change its shape and normal cellular protein (PrPC) misfolds into scrapie prion protein (PrPSc) and becomes abnormal. Scrapie refers to the prion disease first observed in sheep. Scrapie is so named because the sheep behave in other bizarre ways and it is fatal. Human prion diseases have been traditionally classified into kuru, Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler syndrome (GSS), fatal familial insomnia, bovine spongiform encephalopathy (BSE).
Pathogenic Contribution of Prion Diseases
Prion diseases have now become a subject of intense interest because of the risks they pose to public health. In the early 1980s, American neurologist Stanley B. Prusiner and colleagues first identified the ‘proteinaceous infectious particle’. After that, they named it prion. Before his discovery, the diseases now known to be caused by prions were thought to be caused by what were known as “slow viruses”. Stanley Prusiner was awarded the Nobel prize in Medicine in 1997. Prions arise from mutations in the gene that encode the protein. Once prions get inside the brain, they multiply by inducing benign proteins to refold into an abnormal shape. The typical cellular protein structure is thought to consist of several flexible coils called alpha helices. In the prion protein, a conformational transition occurs, and some of these helices are stretched into flat structures called beta-strands. The normal protein conformation is susceptible to protease degradation, whereas the prion protein shape is more resistant to this enzymatic activity. Prions lack nucleic acid i.e., DNA or RNA - which is the genetic material that all other organisms contain. Alzheimer's disease or Parkinson's disease may arise from molecular mechanisms resulting in protein misfolding similar to those that cause prion diseases. The primary symptom of human neurological disorders is dementia, accompanied by motor dysfunction such as cerebellar ataxia and pyramidal or extrapyramidal signs. Prion diseases share significant clinical, neuropathological and cell biological features with another, more common cerebral amyloidosis, Alzheimer’s disease.
Biology of PrPC and PrPSc
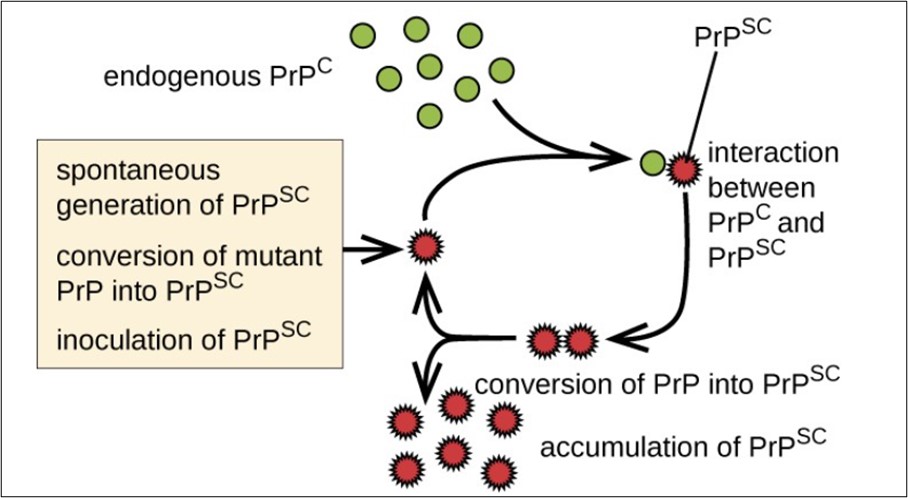
Prion proteins are usually found in healthy brain tissue in a form called PrPC. If this protein gets misfolded into a denatured form (PrPSc), it can cause disease. Normal cellular protein refolds predominantly into beta-pleated sheets and becomes resistant to proteolysis. Thus, PrPSc can induce more PrPC to become misfolded and produce more prion proteins. As PrPSc accumulates, it aggregates and as a consequence, it forms masses of amyloid plaques that give the brain a spongy appearance; that is why these diseases are called spongiform encephalopathy. Thus, as prion proteins multiply, they accumulate within neurons and destroy them. Neuron destruction causes brain tissue to become filled with tiny holes and gives the brain a spongy appearance. The affected person may suffer from memory loss, personality changes, uncoordinated movements and insomnia. These symptoms gradually worsen over time and culminate in coma and death.
There are 6 phenotypes of prion diseases:
i. Scrapie Disease: Scrapie is a fatal, neurodegenerative disease that affects the nervous systems of sheep and goats. The best-studied prion is the scrapie prion, which causes the disease scrapie in sheep. Affected animals lose coordination of their movements, tend to scrape or rub their skin, and eventually cannot walk.
ii. Mad Cow Disease: Initially spread because cattle were fed meals made from all parts of cattle, including brain tissue.
iii. Creutzfeldt-Jakob Disease (CJD) and variant Creutzfeldt-Jakob Disease (vCJD): Sporadic CJD is the most common form of human prion disease.
iv. Familial Fatal Insomnia (FFI): Genetic in origin. It is a rare prion disease that interferes with sleep and leads to deterioration of mental function and loss of coordination.
v. Kuru: Kuru is a fatal degenerative disorder that affects the central nervous system. It is an acquired disorder.
Human prion diseases have become a serious concern as there are currently no effective treatments. No causal therapeutic strategies for human prion diseases have been established to date. Effective treatment of neurodegenerative disease is one of the major challenges. The study on prion diseases following the BSE epidemic has led to these diseases being amongst the best-understood causes of neurodegeneration.